16 Monster cells
This sections produces all the figures used in Supplementary Figure 16 and 17.
# Source setup file
source("./functions/setup.R")
# Load functions
source("./functions/plotHeatmap.R")16.1 Clustering copynumber profiles
First we cluster the copynumber profiles of the previously identified monster cells. Note that the results might slightly differ from the plots in the manuscript due to random seeds being different.
# Load data
annot_p3 = fread("./annotation/P3.tsv", header = F,
col.names = c("library", "section", "pathology"))
profiles_p3 = readRDS("./data/P3_monsters_500kb.rds")
# Run UMAP
set.seed(678) #678
umap_res_p3 = umap(t(profiles_p3[, 4:ncol(profiles_p3)]),
metric = "manhattan",
min_dist = 0,
n_neighbors = 3,
spread = 1,
n_components = 2)
# Transform into dt
umap_dt_p3 = data.table(x = umap_res_p3[, 1],
y = umap_res_p3[, 2],
cell = as.character(1:nrow(umap_res_p3)),
sample = colnames(profiles_p3[, 4:ncol(profiles_p3)]))
# Run clustering and remove cluster 0 (= noise points) if present
clusters_p3 = dbscan::hdbscan(umap_dt_p3[,c(1:2)], minPts = 10)
umap_dt_p3[, cluster := factor(clusters_p3$cluster)]
umap_dt_p3 = umap_dt_p3[cluster != "0", ]
ggplot(umap_dt_p3, aes(x = x, y = y, label = cluster)) +
geom_point(size = 2, alpha = 1, aes(color = cluster)) +
scale_color_npg() +
labs(x = "UMAP 1", y = "UMAP 2") 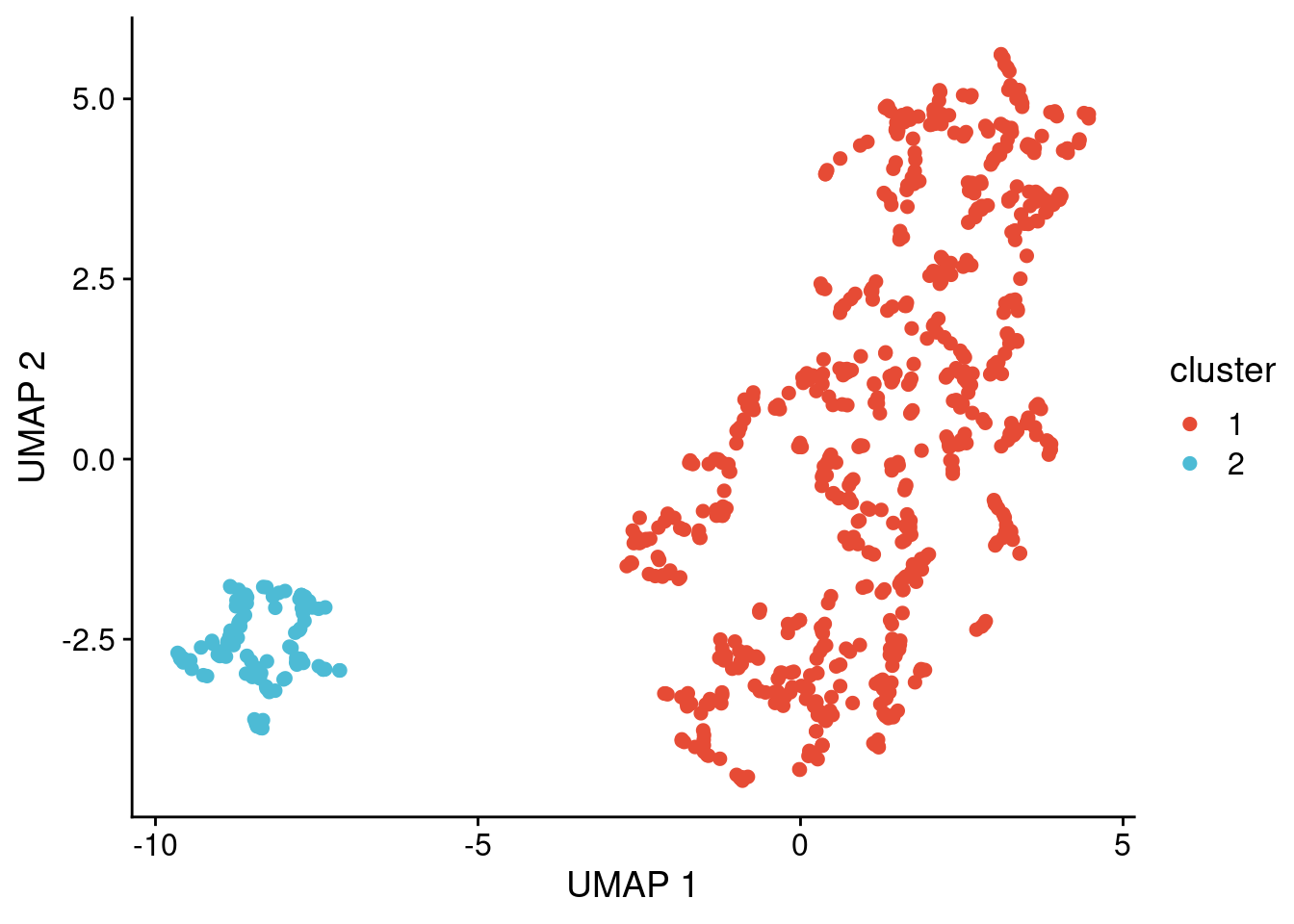
Run the same for P6
# Load data
annot_p6 = fread("./annotation/P6.tsv", header = F,
col.names = c("library", "section", "pathology"))
profiles_p6 = readRDS("./data/P6_monsters_500kb.rds")
# Run UMAP
set.seed(678) #678
umap_res_p6 = umap(t(profiles_p6[, 4:ncol(profiles_p6)]),
metric = "manhattan",
min_dist = 0,
n_neighbors = 3,
spread = 1,
n_components = 2)
# Transform into dt
umap_dt_p6 = data.table(x = umap_res_p6[, 1],
y = umap_res_p6[, 2],
cell = as.character(1:nrow(umap_res_p6)),
sample = colnames(profiles_p6[, 4:ncol(profiles_p6)]))
# Run clustering and remove cluster 0 (= noise points) if present
clusters_p6 = dbscan::hdbscan(umap_dt_p6[,c(1:2)], minPts = 10)
umap_dt_p6[, cluster := factor(clusters_p6$cluster)]
umap_dt_p6 = umap_dt_p6[cluster != "0", ]
ggplot(umap_dt_p6, aes(x = x, y = y, label = cluster)) +
geom_point(size = 2, alpha = 1, aes(color = cluster)) +
scale_color_npg() +
labs(x = "UMAP 1", y = "UMAP 2") 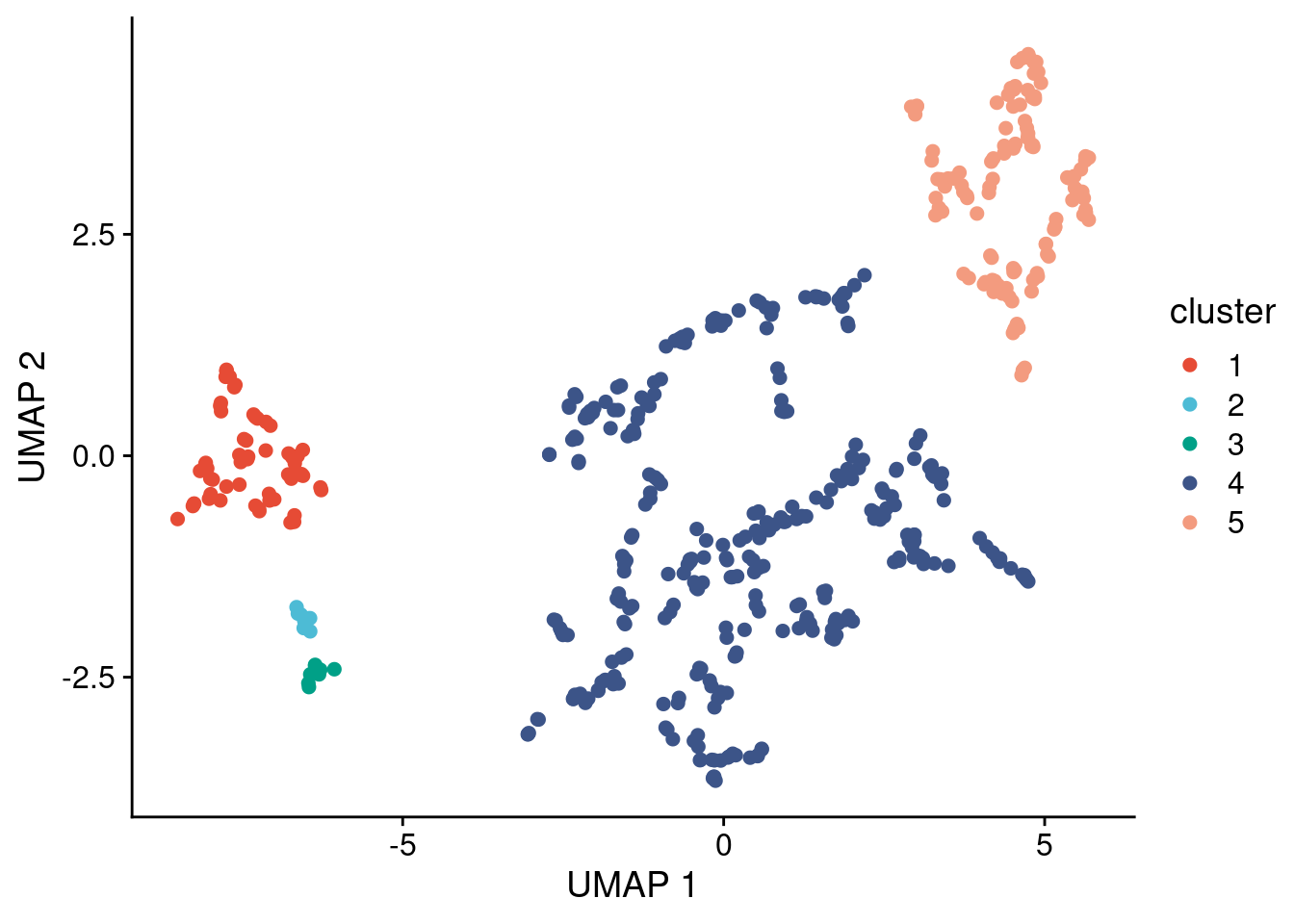
16.2 Plotting genomewide heatmaps
Now we plot, for each cluster, the genomewide heatmap to show the copynumber profiles of these cells.
# For P3
plots = lapply(unique(umap_dt_p3$cluster), function(clust) {
bins = profiles_p3[, 1:3]
profiles = profiles_p3[, umap_dt_p3[cluster == clust, sample], with = FALSE]
plotHeatmap(profiles, bins, dendrogram = FALSE)
})
wrap_plots(plots, ncol = 1, heights = c(0.2, 1))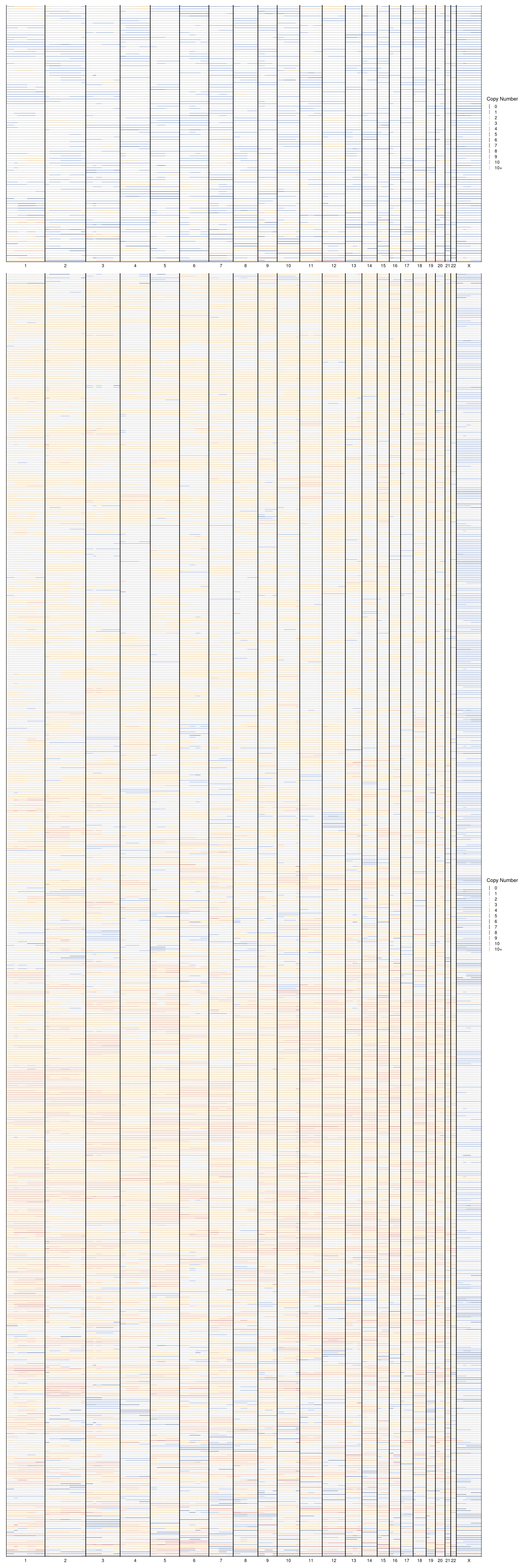
# For P6
plots = lapply(unique(umap_dt_p6$cluster), function(clust) {
bins = profiles_p6[, 1:3]
profiles = profiles_p6[, umap_dt_p6[cluster == clust, sample], with = FALSE]
plotHeatmap(profiles, bins, dendrogram = FALSE)
})
wrap_plots(plots, ncol = 1, heights = c(0.25, 0.6, 0.1, 0.05, 0.05))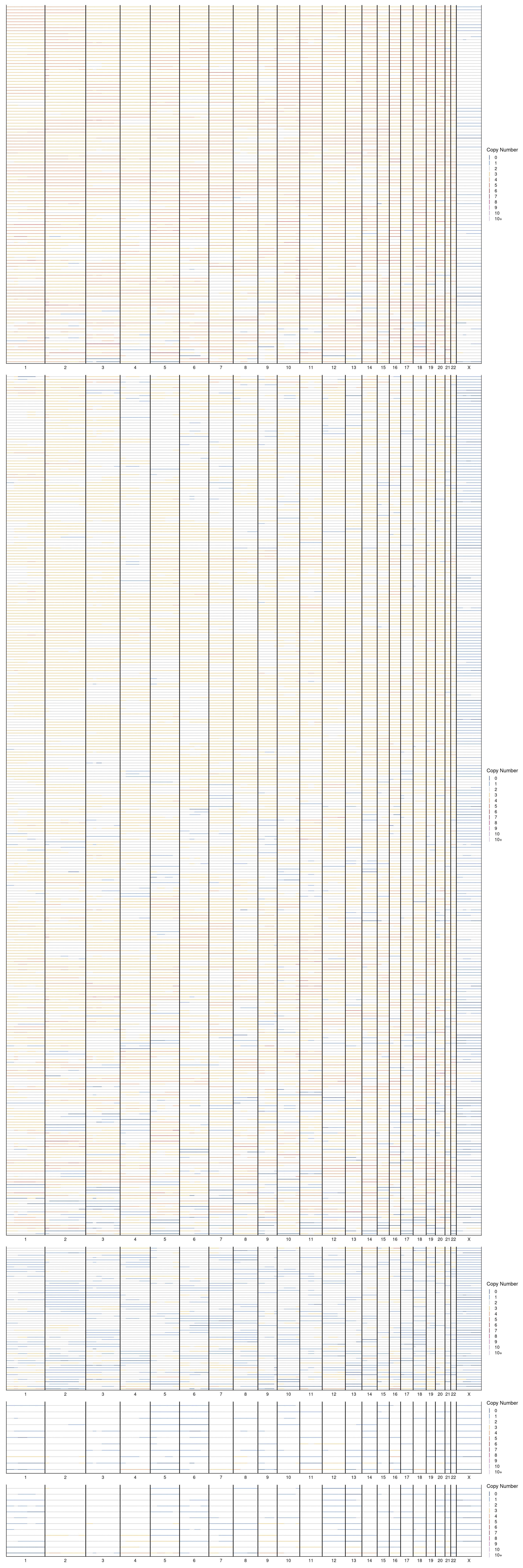
16.3 Plot distribution
Finally, we plot the distribution of where these cells are located in the prostate. First we run it for P3.
locations = umap_dt_p3[, 4:5]
locations[, library := gsub("_.*", "", sample)]
locations = merge(locations, annot_p3, by = "library")
# Get counts and fractions
locations = locations[, .(count = .N), by = .(section, cluster)]
locations = locations[, .(count = count, fraction = count / sum(count), section = section), by = .(cluster)]
# Fill
locations[, x := as.numeric(gsub("L|C.", "", section))]
locations[, y := as.numeric(gsub("L.|C", "", section))]
# Fill NAs
locations = complete(locations, tidyr::expand(locations, x, y))
setDT(locations)
locations[, section := paste0("L", x, "C", y)]
locations = locations[!is.na(cluster), ]
# Plot
ggplot(locations, aes(x = x, y = y, fill = fraction, label = count)) +
geom_tile() +
geom_text() +
facet_wrap(~cluster) +
scale_fill_distiller(name = "Hopeful-Monster\nFraction", palette = "Reds", direction = 1, na.value = "grey") +
geom_hline(yintercept = seq(from = .5, to = max(locations$y), by = 1)) +
geom_vline(xintercept = seq(from = .5, to = max(locations$x), by = 1)) +
scale_y_reverse(expand = c(0, 0), breaks = seq(1, max(locations$y)), labels = seq(1, max(locations$y))) +
scale_x_reverse(expand = c(0, 0), breaks = seq(1, max(locations$x)), labels = seq(1, max(locations$x))) +
theme(axis.title = element_blank())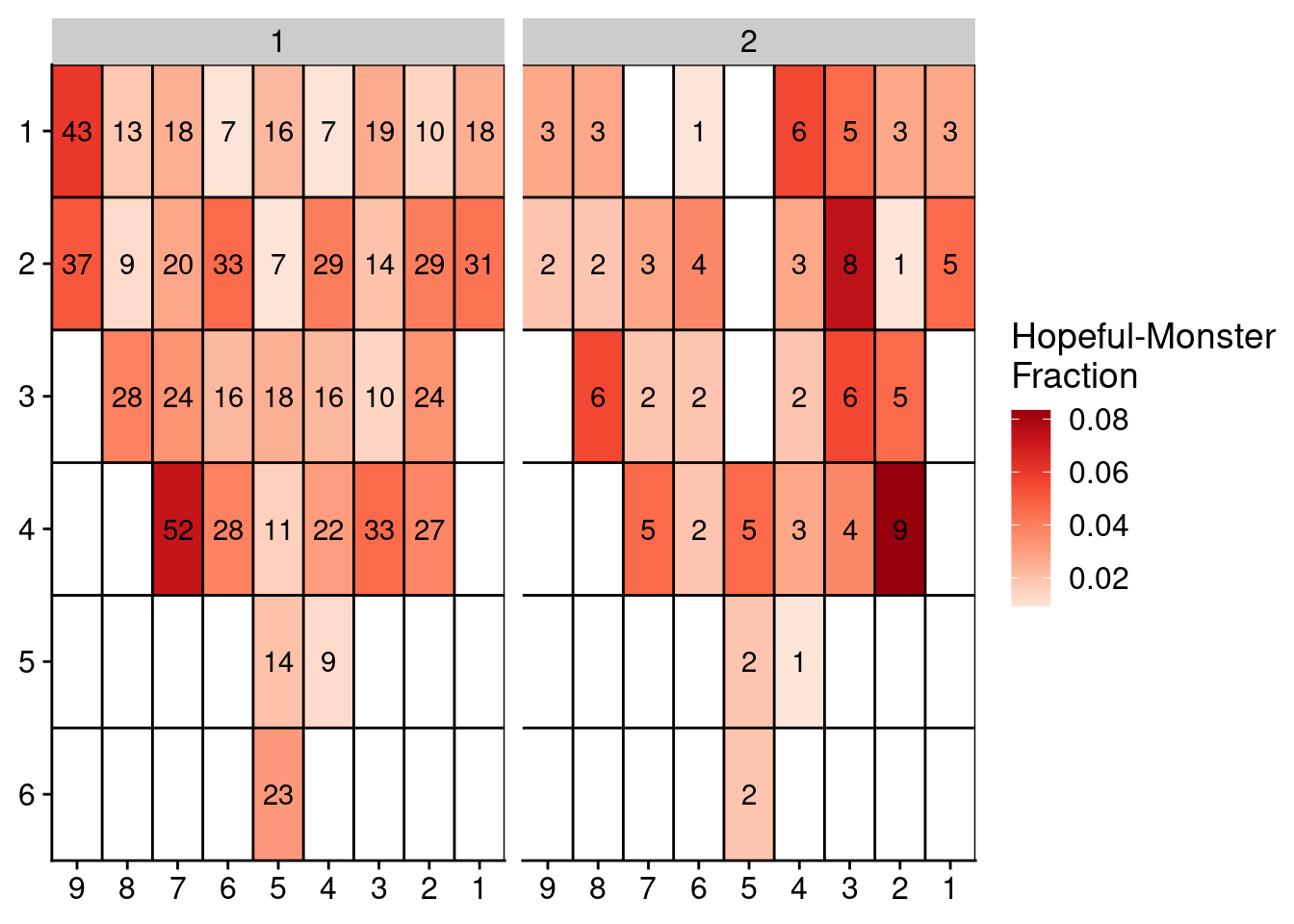
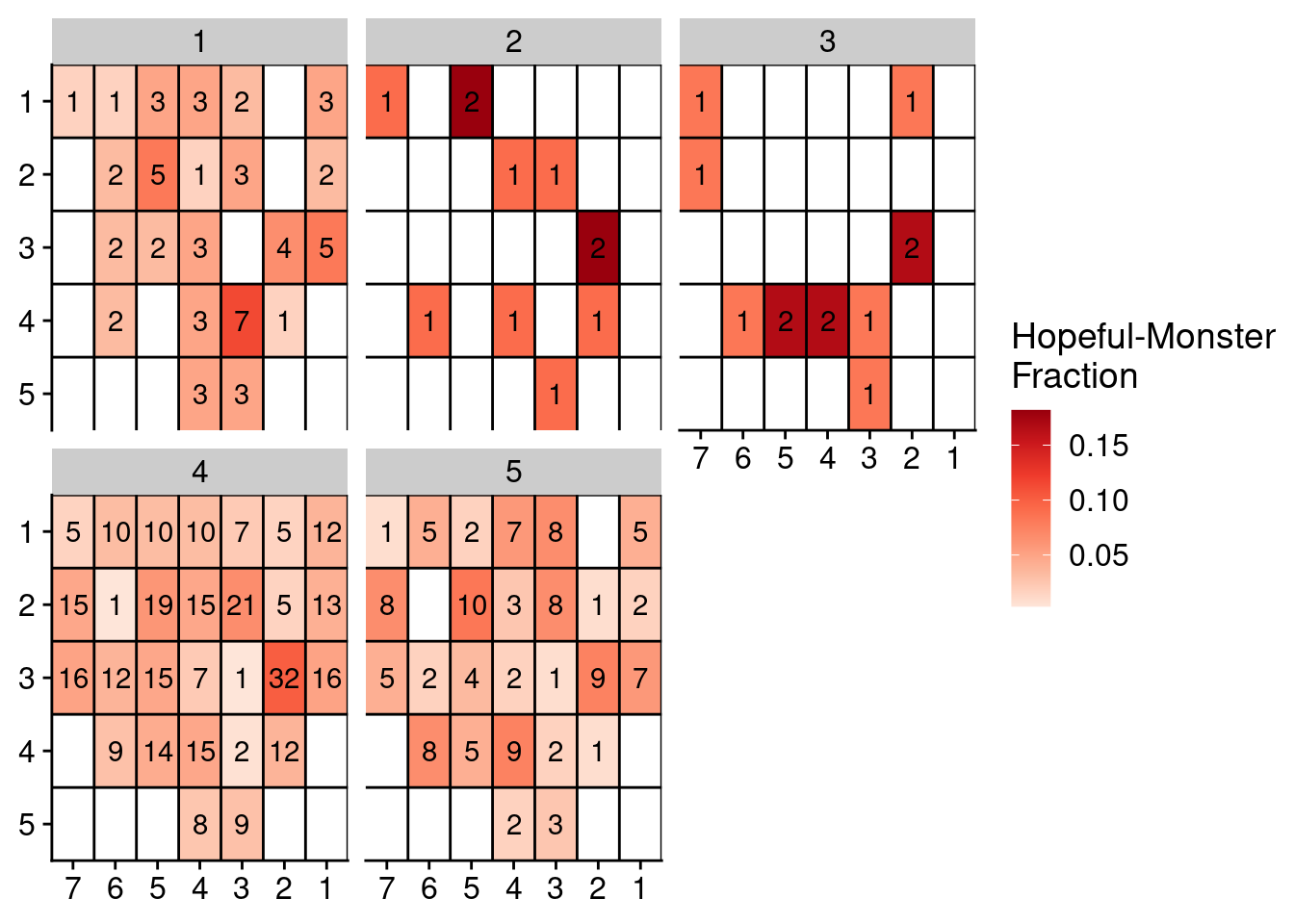
Lastly, we run it for P6.
locations = umap_dt_p6[, 4:5]
locations[, library := gsub("_.*", "", sample)]
locations = merge(locations, annot_p6, by = "library")
# Get counts and fractions
locations = locations[, .(count = .N), by = .(section, cluster)]
locations = locations[, .(count = count, fraction = count / sum(count), section = section), by = .(cluster)]
# Fill
locations[, x := as.numeric(gsub("L|C.", "", section))]
locations[, y := as.numeric(gsub("L.|C", "", section))]
# Fill NAs
locations = complete(locations, tidyr::expand(locations, x, y))
setDT(locations)
locations[, section := paste0("L", x, "C", y)]
locations = locations[!is.na(cluster), ]
# Plot
ggplot(locations, aes(x = x, y = y, fill = fraction, label = count)) +
geom_tile() +
geom_text() +
facet_wrap(~cluster) +
scale_fill_distiller(name = "Hopeful-Monster\nFraction", palette = "Reds", direction = 1, na.value = "grey") +
geom_hline(yintercept = seq(from = .5, to = max(locations$y), by = 1)) +
geom_vline(xintercept = seq(from = .5, to = max(locations$x), by = 1)) +
scale_y_reverse(expand = c(0, 0), breaks = seq(1, max(locations$y)), labels = seq(1, max(locations$y))) +
scale_x_reverse(expand = c(0, 0), breaks = seq(1, max(locations$x)), labels = seq(1, max(locations$x))) +
theme(axis.title = element_blank())