15 Individual subclone distribution in prostate
This sections produces all the figures used in Supplementary Figure 11, 12, 13 and 14.
# Source setup file
source("./functions/setup.R")
# Load functions
source("./functions/plotHeatmap.R")
source("./functions/plotProfile.R")15.1 Plotting individual subclone distribution
Here we plot, for each detected subclone, the distribution within the prostate. The scripts to obtain the list of subclones and the associated entropy is in the Rmd file for Figure 2. Once again, we do this first for P3.
# Load data
profiles = readRDS("./data/P3_pseudodiploid_500kb.rds")
clones = fread("./data/subclones/P3_clones.tsv")
annot = fread("./annotation/P3.tsv", header = F)
entropy = fread("./data/entropy/P3_entropy.tsv")
# Make copy of clones
clones_annot = copy(clones)
# Prepare cn profiles
profiles = profiles[, c("chr", "start", "end", clones$sample_id), with = F]
# Melt into long format and merge with clone information
profiles = melt(profiles, id.vars = c("chr", "start", "end"))
profiles = merge(profiles, clones, by.x = "variable", by.y = "sample_id")
# Get consensus profiles
profiles = profiles[, .(cn = round(median(value))), by = .(chr, start, end, cluster)]
profiles = dcast(profiles, chr+start+end ~ cluster, value.var = "cn")
profiles = profiles[gtools::mixedorder(profiles$chr)]
# Merge clones with annotation data
clones[, library := gsub("_.*", "", sample_id)]
clones = merge(clones, annot, by.x = "library", by.y = "V1", all.y = TRUE)
setnames(clones, "V2", "section")
# Get fraction of each subclone in each section
clones = clones[, .N, by = .(cluster, section)]
clones = clones[, .(count = N, section = section, fraction = N / sum(N)), by = .(cluster)]
# Add coordinates
clones[, x := as.numeric(gsub("L|C.", "", section))]
clones[, y := as.numeric(gsub("L.|C", "", section))]
# Fill NAs
clones[, section := paste0("L", x, "C", y)]
clones = clones |> complete(section, cluster)
setDT(clones)
# Readd coordinates
clones[, x := as.numeric(gsub("L|C.", "", section))]
clones[, y := as.numeric(gsub("L.|C", "", section))]
# Loop through clones and combine the distribution heatmap and the genomewide heatmap
plots = lapply(entropy$clone, function(clone) {
# Plot profile heatmap
heatmap = plotHeatmap(profiles[, clone, with = F], profiles[, 1:3], linesize = 20, dendrogram = F, order = clone, rast = T) + theme(legend.position = "none")
# Complete
dt = clones[cluster == clone]
plt =
ggplot(dt, aes(x = x, y = y, fill = fraction, label = count)) +
geom_tile() +
geom_text() +
labs(title = clones_annot[cluster == clone, name]) +
scale_fill_distiller(name = "Subclone\nFraction", palette = "Reds", direction = 1, na.value = "grey", limits = c(0, 1)) +
geom_hline(yintercept = seq(from = .5, to = max(dt$y), by = 1)) +
geom_vline(xintercept = seq(from = .5, to = max(dt$x), by = 1)) +
scale_y_reverse(expand = c(0, 0), breaks = seq(1, max(dt$y)), labels = seq(1, max(dt$y))) +
scale_x_reverse(expand = c(0, 0), breaks = seq(1, max(dt$x)), labels = seq(1, max(dt$x))) +
theme(axis.title = element_blank())
# Combine plots
combined = wrap_plots(heatmap, plt, ncol = 1, heights = c(0.125, 1))
return(combined)
})
# Plot everything at once
wrap_plots(plots, ncol = 4) # You can also plot 1 by 1 by not running this but simply running 'plots'
And now for P6
# Load data
profiles = readRDS("./data/P6_pseudodiploid_500kb.rds")
clones = fread("./data/subclones//P6_clones.tsv")
annot = fread("./annotation/P6.tsv", header = F)
entropy = fread("./data/entropy/P6_entropy.tsv")
# Make copy of clones
clones_annot = copy(clones)
# Prepare cn profiles
profiles = profiles[, c("chr", "start", "end", clones$sample_id), with = F]
# Melt into long format and merge with clone information
profiles = melt(profiles, id.vars = c("chr", "start", "end"))
profiles = merge(profiles, clones, by.x = "variable", by.y = "sample_id")
# Get consensus profiles
profiles = profiles[, .(cn = round(median(value))), by = .(chr, start, end, cluster)]
profiles = dcast(profiles, chr+start+end ~ cluster, value.var = "cn")
profiles = profiles[gtools::mixedorder(profiles$chr)]
# Merge clones with annotation data
clones[, library := gsub("_.*", "", sample_id)]
clones = merge(clones, annot, by.x = "library", by.y = "V1", all.y = TRUE)
setnames(clones, "V2", "section")
# Get fraction of each subclone in each section
clones = clones[, .N, by = .(cluster, section)]
clones = clones[, .(count = N, section = section, fraction = N / sum(N)), by = .(cluster)]
# Add coordinates
clones[, x := as.numeric(gsub("L|C.", "", section))]
clones[, y := as.numeric(gsub("L.|C", "", section))]
# Fill NAs
clones[, section := paste0("L", x, "C", y)]
clones = clones |> complete(section, cluster)
setDT(clones)
# Readd coordinates
clones[, x := as.numeric(gsub("L|C.", "", section))]
clones[, y := as.numeric(gsub("L.|C", "", section))]
# Loop through clones and combine the distribution heatmap and the genomewide heatmap
plots = lapply(entropy$clone, function(clone) {
# Plot profile heatmap
heatmap = plotHeatmap(profiles[, clone, with = F], profiles[, 1:3], linesize = 20, dendrogram = F, order = clone, rast = T) + theme(legend.position = "none")
# Complete
dt = clones[cluster == clone]
plt =
ggplot(dt, aes(x = x, y = y, fill = fraction, label = count)) +
geom_tile() +
geom_text() +
labs(title = clones_annot[cluster == clone, name]) +
scale_fill_distiller(name = "Subclone\nFraction", palette = "Reds", direction = 1, na.value = "grey", limits = c(0, 1)) +
geom_hline(yintercept = seq(from = .5, to = max(dt$y), by = 1)) +
geom_vline(xintercept = seq(from = .5, to = max(dt$x), by = 1)) +
scale_y_reverse(expand = c(0, 0), breaks = seq(1, max(dt$y)), labels = seq(1, max(dt$y))) +
scale_x_reverse(expand = c(0, 0), breaks = seq(1, max(dt$x)), labels = seq(1, max(dt$x))) +
theme(axis.title = element_blank())
# Combine plots
combined = wrap_plots(heatmap, plt, ncol = 1, heights = c(0.125, 1))
return(combined)
})
# Plot everything at once
wrap_plots(plots, ncol = 4) # You can also plot 1 by 1 by not running this but simply running 'plots'
Now, for the top 5 most local (lowest entropy) subclones, we plot the individual copy number profiles for manual inspection.
# Load data
cnv = readRDS("./data/P3_cnv.rds")
clones = fread("./data/subclones/P3_clones.tsv")
entropy = fread("./data/entropy/P3_entropy.tsv")
# Select top 5 clones
top5 = entropy[1:5, name]
# Select profiles
top_profiles = clones[name %in% top5, sample_id]
# Loop through profiles and plot
plots = lapply(top_profiles, function(x) {
plt = plotProfile(cnv$copynumber[[x]], cnv$counts_gc[[x]] * cnv$ploidies[sample == x, ploidy], bins = cnv$bins) +
ggtitle(paste0(clones[sample_id == x, name], " - ", x))
return(plt)
})
wrap_plots(plots, ncol = 3)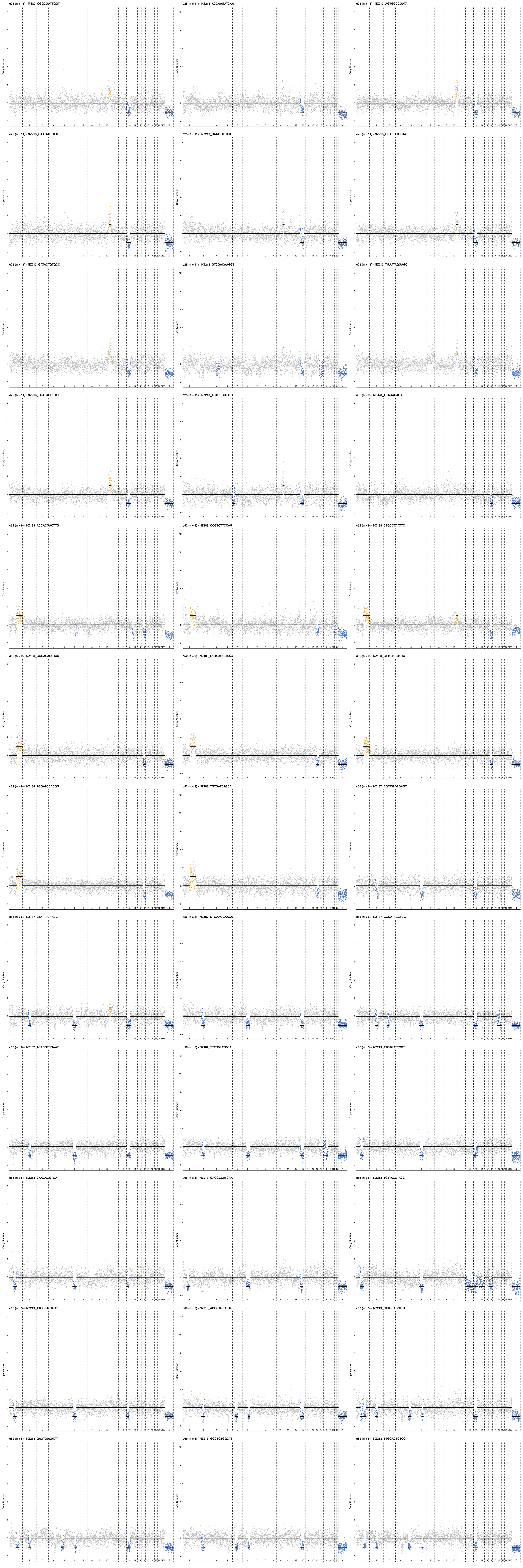
And repeat for P6.
# Load data
cnv = readRDS("./data/P6_cnv.rds")
clones = fread("./data/subclones/P6_clones.tsv")
entropy = fread("./data/entropy/P6_entropy.tsv")
# Select top 5 clones
top5 = entropy[1:5, name]
# Select profiles
top_profiles = clones[name %in% top5, sample_id]
# Loop through profiles and plot
plots = lapply(top_profiles, function(x) {
plt = plotProfile(cnv$copynumber[[x]], cnv$counts_gc[[x]] * cnv$ploidies[sample == x, ploidy], bins = cnv$bins) +
ggtitle(paste0(clones[sample_id == x, name], " - ", x))
return(plt)
})
wrap_plots(plots, ncol = 3)